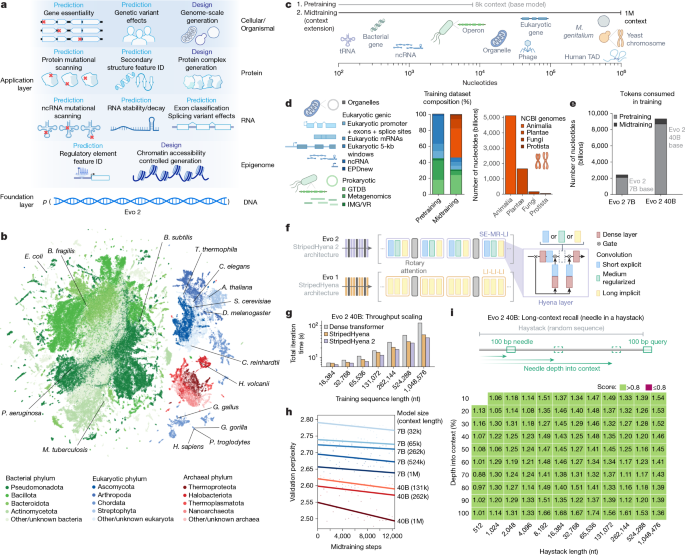
Era il marzo 2025 quando apparve in preprint il primo articolo che descriveva Evo 2, un modello di intelligenza artificiale addestrato su 128.000 genomi raccolti da ogni angolo dell’albero della vita: dagli esseri umani ai batteri unicellulari più antichi (ne avevamo parlato ampiamente qui). Esattamente un anno dopo, il 4 marzo 2026, quella ricerca è approdata sulle pagine di Nature, aggiornata, arricchita di nuovi esperimenti e accompagnata da uno strumento inedito — Evo Designer — che permette a qualsiasi laboratorio del mondo di generare sequenze di DNA in tempo reale, a partire da semplici istruzioni testuali.
Addestrato su 9,3 trilioni di nucleotidi provenienti da oltre 128.000 specie, Evo 2 è oggi il più grande modello di intelligenza artificiale completamente open source del suo genere finora sviluppato. La risorsa è stata resa disponibile gratuitamente dai suoi co-creatori, tramite GitHub dell’istituto di ricerca no-profit Arc Institute e tramite il framework BioNeMo dell’azienda tecnologica statunitense Nvidia.
In dodici mesi, Evo 2 ha ricevuto citazioni scientifiche, totalizzato più di 88.000 download e registrato più di 8 milioni di richieste tramite API. Gruppi di ricerca a Yale, Johns Hopkins, North Carolina State e University of Washington lo hanno già messo al lavoro su problemi concreti: dalla previsione del rischio di Alzheimer all’analisi della struttura tridimensionale del genoma. Non è più uno strumento sperimentale. È diventato infrastruttura scientifica.
Nel 2025, Brian Hie e i suoi colleghi avevano già usato versioni precedenti di Evo per scrivere genomi di fagi — virus che infettano i batteri. Su 285 sequenze generate artificialmente e inserite in cellule di Escherichia coli, 16 avevano prodotto virus funzionanti, capaci di uccidere i batteri ospiti. Un tasso di successo del 5-6% che, considerata la complessità del problema, la comunità scientifica aveva accolto con sorpresa e interesse. I fagi rimangono sistemi molto più semplici di un batterio vero, ma dimostrano che l’IA può già oggi produrre sistemi biologici funzionali partendo da zero.
Scrivere nel genoma come su un foglio bianco
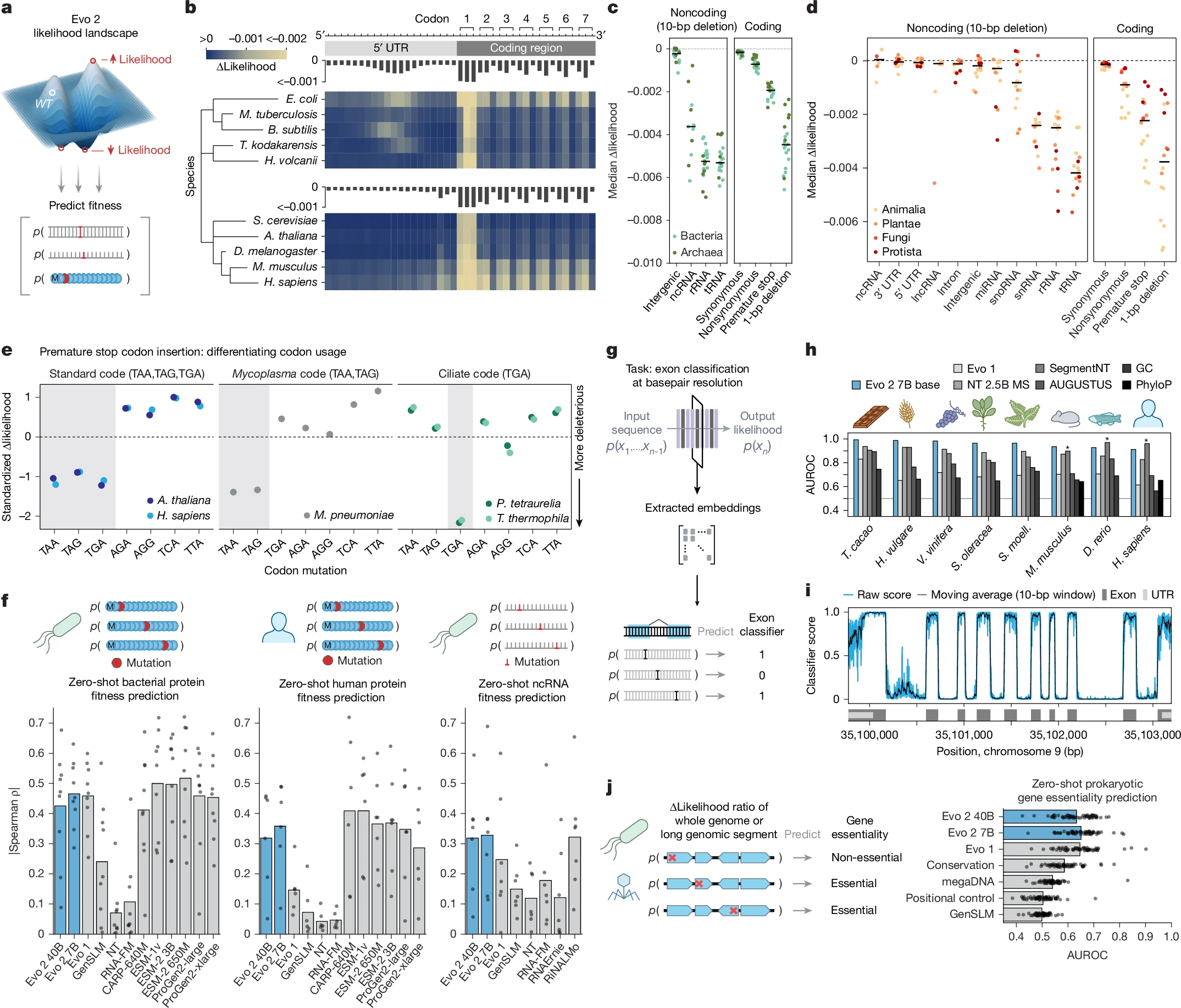
La novità scientifica più rilevante della pubblicazione aggiornata su Nature riguarda la capacità di Evo 2 di progettare sequenze di DNA sintetico lunghe fino a 20.000 nucleotidi — le cosiddette sequenze a lungo raggio — e di farlo con un’efficacia sorprendentemente alta. Il team guidato da Patrick Hsu e Brian Hie all’Arc Institute di Palo Alto ha dimostrato sperimentalmente che queste sequenze artificiali riescono a modificare l’accessibilità della cromatina, ovvero la struttura tridimensionale con cui il DNA si avvolge all’interno del nucleo cellulare, con una precisione misurabile. I test eseguiti su cellule vere — sia di topo che umane — hanno restituito valori di accuratezza compresi tra 0,92 e 0,95 su scala AUROC, un risultato che in biologia computazionale si considera eccellente.
In termini più concreti: il modello non si limita a leggere e interpretare il genoma come un testo, ma impara a riscriverne frammenti producendo effetti biologici reali e prevedibili. Patrick Hsu ha descritto sui social come il suo laboratorio abbia persino usato Evo 2 per scrivere messaggi in codice Morse all’interno dell’epigenoma — una dimostrazione volutamente spettacolare della capacità del modello di controllare la struttura del DNA oltre la semplice sequenza di basi.
Il sogno del genoma sintetico
Ma la domanda che rimbalza nella comunità scientifica è un’altra: Evo 2 è già in grado di scrivere un genoma completo che funzioni all’interno di una cellula viva? La risposta, per ora, è no — o almeno non ancora. Il team ha pubblicato tre tentativi ambiziosi: un genoma ispirato a quello di Mycoplasma genitalium — uno dei batteri con il corredo genetico più semplice in natura — la sequenza del DNA mitocondriale umano e un cromosoma di lievito. Le simulazioni computazionali hanno suggerito che circa il 70% dei geni presenti nelle sequenze generate sembrano realistici. Un risultato notevole per un modello linguistico. Ma non sufficiente per la vita.
“Non si può progettare la vita al 70%”, ha detto senza mezzi termini a Nature Nico Claassens, biologo sintetico all’Università di Wageningen nei Paesi Bassi. “Puoi farlo al computer, ma non sarà funzionale.” Anche un solo gene essenziale mancante o mal codificato è sufficiente a rendere sterile l’intero progetto. E non si tratta solo di avere tutti i geni giusti: conta anche l’ordine in cui sono disposti lungo il cromosoma, le interazioni regolatorie tra regioni distanti, la geometria dello spazio nucleare. Fattori che nessun modello linguistico — per quanto potente — ha ancora imparato a padroneggiare del tutto.
Un ulteriore elemento critico è emerso questa settimana da un preprint dell’Università del Texas ad Austin: i genomi generati da Evo 2 presentano un’organizzazione strutturalmente diversa da quella dei genomi naturali e mancano di alcune caratteristiche chiave. I ricercatori texani non escludono che queste sequenze possano risultare funzionali, ma segnalano che difficilmente potranno offrire intuizioni genuine sull’evoluzione genomica — uno degli obiettivi originari che aveva motivato l’intera impresa della genomica sintetica.
Il collo di bottiglia non è più l’algoritmo
Brian Hie è convinto (chiaramente) che la qualità delle sequenze generate migliorerà rapidamente, ma ammette che il vero problema si sta spostando altrove. Sintetizzare fisicamente un genoma — assemblare centinaia di migliaia di nucleotidi nell’ordine corretto — ha costi enormi e richiede tecnologie che non stanno evolvendo alla stessa velocità dei modelli di intelligenza artificiale. “Gli esperimenti stanno rapidamente diventando il collo di bottiglia”, ha dichiarato. “A questa scala, ci scontriamo con il costo della sintesi del DNA e il costo della sua assemblaggio.”
La visione che emerge dal gruppo dell’Arc Institute per superare questo ostacolo è quella dei laboratori autonomi: sistemi in cui intelligenza artificiale e robotica collaborano in cicli continui di progettazione, sintesi, test e raffinamento di piccoli segmenti genomici, che potrebbero essere successivamente assemblati in un genoma completo. Un approccio modulare, quasi ingegneristico, che ricorda più la costruzione di un edificio — mattone per mattone — che la scrittura di un libro dall’inizio alla fine.

{kind=link}
{kind=link}